keywords: ALS variants, regional ALS, brachial amyotrophic diplegia, amyotrophic leg diplegia, flail arm ALS
Purpose: We illustrate three patients with regional amyotrophic lateral sclerosis (ALS) variants and hope to improve accuracy in diagnosis for this scarce group of diseases.
Case report: Amyotrophic lateral sclerosis (ALS) represents a broad spectrum of acquired and inherited neurodegenerative conditions involving the upper and motor neurons. Typical ALS remains a clinical diagnosis that is not hard to diagnose. Still, when it comes to atypical forms of ALS, the physicians may face some difficulties differentiating between atypical forms of ALS and other neurological diseases, such as multifocal motor neuropathy, chronic inflammatory demyelinating polyneuropathy, and spinal muscular atrophy. Both brachial amyotrophic diplegia (BAD) and leg amyotrophic diplegia (LAD) are considered regional variants of ALS. We are here to report two cases of BAD and one case of LAD. All these 3 cases showed progression of the disease after longitudinal follow- up for approximately two years. However, after two years, their disease progressions were slow and confined to their 'regions' of upper or lower limbs.
Conclusion: BAD and LAD are unique regional variants of ALS with a significantly better prognosis than typical ALS. The phenotypic characteristics of regional ALS variants must be recognized when physicians are to tailor advice on disease progression, disease outcome, drug therapy, and end-of-life planning for patients with ALS or ALS variants.
Keywords: ALS variants, regional ALS, brachial amyotrophic diplegia, amyotrophic leg diplegia, flail arm ALS
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) includes a heterogeneous expanding group of disorders involving the upper and lower motor neurons (1), which is reflected in the Revised El Escorial (2) and Awaji (3) criteria. Patients with ALS typically have a combination of both upper and lower motor neuron signs that affect multiple segments of the body. To date, diagnosis of MND is based on the combination of classical presentation, examination findings, electrophysiological features, and exclusion of other potential causes. Typical ALS is not hard to diagnose, but when it comes to the atypical form of MND, the physicians may face some difficulties in differentiating clearly between atypical forms of motor neuron disease (MND) and other neurological diseases, such as multifocal motor neuropathy, chronic inflammatory demyelinating polyneuropathy, spinal muscular atrophy, and cervical myelopathy.
Typical ALS is characterized by a rapidly progressive, invariably fatal disease, comprised of mixed upper and lower motor neuron involvement in different spinal cord regions, with overall median survival of 3 to 4 years (4). The main four subtypes for typical ALS include classic limb onset ALS, progressive bulbar palsy (bulbar onset ALS), primary lateral sclerosis (PLS), and a lower motor neuron dominant variant called progressive muscular atrophy (PMA). Apart from the four main subtypes of ALS, other forms have been recognized.
Another two forms of ALS variants have been recognized since the late 19th century but relatively inadequately studied as it was extremely rare. These being the brachial amyotrophic diplegia and leg amyotrophic diplegia. Both brachial amyotrophic diplegia and leg amyotrophic diplegia are considered regional variants of ALS. The regional variants of ALS herald a better prognosis than typical ALS (5). Brachial amyotrophic diplegia, also known as flail arm syndrome, manifests as slowly progressive loss of arm function, often initially proximal and asymmetric. Leg amyotrophic diplegia, also known as flail leg syndrome, involves slowly progressive loss of leg function. It is also slowly progressive and asymmetric but may be proximal or distal initially (5). To understand the natural history and presentation of these extremely rare ALS variants, we report 3 cases of regional variants of ALS.
RESULT
Case 1: Brachial amyotrophy diplegia (BAD)
A 47 years old man presented with progressive bilateral upper limbs weakness for a year. There were no associated sensory symptoms. His weakness started in the right shoulder and progressed to involve the left shoulder. He started to have difficulty in carrying heavy objects six months from the onset of his symptoms. His weakness spread over to affect the proximal arms. There were no swallowing and speech difficulties. He denied weakness of his lower limbs.
On examination, there are significant muscles wasting over both supraspinatus and infraspinatus muscles, both deltoids and biceps muscles [Figure 1]. There was no wasting or fasciculation of the tongue and lower limbs muscles. No gynecomastia was noted. There was bilateral shoulder girdle weakness. Both shoulder abduction and adduction power were 3/5 according to Medical Research Council Scale for Muscle Strength (MCR scale). Both elbow flexion and extension power were 4/5. Motor power of the wrist flexion and extension, the neck flexion and extension were normal. Motor power in the lower extremities was also normal. Sensation to both pinprick and proprioception was normal. Muscle stretch reflexes were reduced in the upper extremities and normal in the lower extremities.
Blood investigation showed a normal creatine kinase (CK) level. Anti GM-1 antibody was normal. Magnetic resonance imaging (MRI) of the brain and cervical spine were normal. His nerve conduction study showed a normal sensory nerve conduction study. Motor nerve conduction study shows diffusely reduced compound motor action potentials (CMAPs) over both upper limbs, sparing the lower limbs. No conduction block was recorded in the study. Needle electromyography (EMG) of multiple upper and lower limbs muscles, cervical and thoracic paravertebral muscles, and the tongue muscles showed chronic neurogenic changes with active denervation over left biceps, both deltoid, and biceps, right C8, T1, T12 paravertebral muscles. Spontaneous fasciculations are recorded over the right biceps and left deltoid muscles. There was sparing of the lower limb muscles. Transcranial magnetic stimulation shows normal central motor conduction time (CMCTs) and motor evoked potentials (MEPs) to both upper limbs and left lower limb.
Over the next one-year follow-up, his weakness in both upper limbs progressed to involve both proximal and distal upper extremities muscles. His lower limbs' power, respiratory muscle, and bulbar muscles function remained normal.
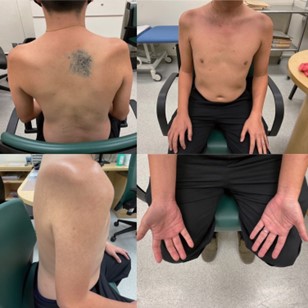
Figure 1. Severe muscle wasting over both supraspinatus and infraspinatus muscles, both deltoids and biceps muscles in our patient with brachial amyotrophic diplegia.
Case 2: Leg amyotrophy diplegia (LAD)
A 59 years old man was referred for five months history of right thigh wasting. There was no weakness and numbness. Initial examination revealed wasting of the right quadriceps muscles. The rest of the neurological examination was unremarkable. His serum creatinine kinase (CK) level and HbA1c were normal. The cervical and lumbar spine MRI showed mild L5 spondylolisthesis. There was no foraminal stenosis or cord compression. His nerve conduction study showed mildly reduced right tibial and peroneal CMAP. The sensory nerve conduction study was unremarkable.
He was followed up closely for the next two years. He started to develop weakness in the right lower limb six months later (from the first consultation). The weakness progressed to involve both lower extremities 12 months later (from his first consultation). On examination, asymmetrical lower limb wasting was more prominent over the right lower limb [Figure 2]. Fasciculation was noted over the right quadriceps. Muscle strength testing showed the right hip flexion and extension power 3/5, right knee flexion and extension were 4/5, the left hip flexion was 4/5. The rest of the motor power was 5/5. Sensory examination of pinprick, proprioception and vibration sensation was normal. Muscle stretch reflexes were normal. There was no gynaecomastia, no tongue wasting, or fasciculation.
Nerve conduction study showed reduced right peroneal and tibial CMAPs. Needle electromyography showed chronic neurogenic changes with active denervation over right and left tibialis anterior, right gastrocnemius, right vastus lateralis. Needle EMG of the right EDC, right and left biceps, left paravertebral C5, T7, and T12 muscles are within normal limits. Spontaneous fasciculation potentials were recorded over the right vastus lateralis and right tibialis anterior muscles. Transcranial magnetic stimulation shows normal central motor conduction time (CMCTs) and motor evoked potentials (MEPs) to both upper limbs and left lower limb.
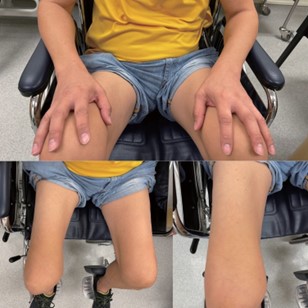
Figure 2. Asymmetrical lower limb wasting (more prominent over the right lower limb) in our patient with leg amyotrophic diplegia. There was sparing of the upper limb muscles.
Case 3: Leg amyotrophy diplegia (LAD)
A 60-year-old lady presented with a one-year history of progressive weakness in the right lower limb, followed by a two-month history of left lower limb weakness. There was neither numbness nor slurred speech. Both bowel movements and urination were normal.
On examination, asymmetrical lower limb wasting was more prominent over the right lower limb [Figure 3]. No fasciculation was noted. The muscle tone of both lower limbs was normal. Muscle stretch reflexes were normal. The motor power of the right big toe extension was 0/5, while the motor power of the right hip flexion and right ankle dorsiflexion was 3/5. The rest of the lower limbs' motor power was 4/5. Sensory examination of pinprick, proprioception and vibration sensation was normal. There was no tongue fasciculation. Examination of both upper limbs was unremarkable.
Blood investigations of serum creatinine kinase, serum Vitamin B12 level, folate, calcium, phosphate, serum protein electrophoresis, HIV, HTLV, paraneoplastic antibody, ANA, ENA, dsDNA, Hepatitis B, and C screening are unremarkable. Her lumbar, thoracic, and cervical spine MRI showed no significant spinal canal, lateral recess, or foraminal exit stenosis. Her sensory nerve conduction study (NCS) was normal. Motor NCS showed no conduction blocks. There was diminished right deep peroneal, right tibial, and bilateral peroneal compound muscle action potentials (CMAPs), sparing the upper limb muscles. Needle EMG study showed chronic neurogenic changes in bilateral tibialis anterior, bilateral vastus lateralis, and right biceps femoris muscles with active denervation in the bilateral tibialis anterior, bilateral vastus lateralis, right biceps femoris, right gastrocnemius, and right L4 and right T9 paraspinal muscles.
Upon her follow-up, two years from the onset of her weakness, there was a deterioration of her lower limbs weakness and wasting with fasciculation noted over bilateral thighs. Her upper limbs' power, respiratory muscles, bulbar muscle functions remained intact.
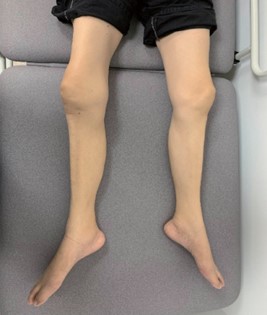
Figure 3. Asymmetrical lower limb wasting (more prominent over the right lower limb) in our patient with leg amyotrophic diplegia.
DISCUSSION
Amyotrophic lateral sclerosis (ALS) is a heterogenous disease with a variety of potential clinical phenotypes. It remains a clinical diagnosis without a unique marker. Patients with ALS typically have a combination of both upper and lower motor neuron signs that affect multiple segments of the body (1). The four main clinical categories of ALS were classic limb onset ALS, progressive bulbar palsy (bulbar onset ALS), primary lateral sclerosis (PLS), and the lower motor neuron form termed progressive muscular atrophy (PMA).
Apart from these four main subtypes of ALS, other forms have been recognized. Regional ALS variants have been described where the disease is restricted to one spinal region at presentation and isolated to a single spinal region for many years. These being the brachial amyotrophic diplegia (BAD) and leg amyotrophic diplegia (LAD) (4). In this case series, which consists of one patient with BAD, and two patients with LAD, the patients were followed up for 1.5 to two years. The disease is still limited to the same body segment throughout the follow-up, with no upper motor neuron signs. All these 3 cases showed progression of the disease after longitudinal follow- up for approximately two years. However, their disease progressions were slow.
In general, both BAD and LAD portend a better prognosis than classic ALS. Over time, some patients with regional variants of ALS will progress to more widespread disease, affecting multiple segments of the body. To date, the exact proportion of patients that convert to widespread disease and the factors that predict progression remain somewhat uncertain (5). The previous study has suggested that for all ALS phenotypes, a longer duration to the involvement of the second region was associated with a better prognosis (7).
Brachial amyotrophic diplegia, also known as flail arm syndrome, manifests as slowly progressive loss of arm function, often initially proximal and asymmetric (5). Our first case with the BAD presentation was typical for BAD, where it was initially proximal, then slowly progressed to involve distally and contralateral upper limb in an asymmetric pattern. On the other hand, leg amyotrophic diplegia usually presents with a slowly progressive loss of leg function. It manifests as asymmetric, slowly progressive, and can be proximal or distal initially. Our second case with LAD presented with slowly progressive, asymmetric proximal lower limb weakness, while the third case presented with slowly progressive, asymmetric proximal, and distal lower limb weakness.
As compared with the previous large cohort study in London and Melbourne by Wijesekera LC et al. (8), our cohort shared some similarities with this large cohort study. All the three patients fulfilled the same operational definitions for the flail arms syndrome (BAD) and flail legs syndrome (LAD). The functional involvement of our three patients were confined to the flail limbs for at least 12 months after onset of symptoms (to differentiate regional ALS variants from early-onset ALS or PMA). In the London cohort, the mean age of onset for BAD and LAD were 57.3 and 55.0, respectively, while the mean age of onset in the Melbourne cohort for BAD and LAD were 61.9 and 58.1, respectively. The age of onset was similar with our three patients with BAD or LAD (mean age of onset is 55.3).
Wijesekera LC et al. also confirmed that BAD is more common in males, with a male-to-female ratio of 4:1. For LAD, the male-to-female ratio in the London cohort was 1:1. Our cohort consists of only three Asian patients (one male BAD patient, one male and one female LAD patients). It is difficult to compare our finding from the predominantly Caucasian cohort in London and Melbourne by Wijesekera LC et al. in view of the small cohort size. In the future, large population-based samples will be required to compare the similarities and differences between Caucasian and Asian patients with regional ALS variants.
To date, no single diagnostic test is available for ALS and its variants (9). Diagnosis is based on the combination of history taking, physical examination, electrophysiologic findings, and exclusion of other potential differential diagnoses. Nerve conduction study should demonstrate sparing of the sensory study. Sensory nerve involvement should suggest other differentials, such as polyneuropathy, vasculitis, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), and Kennedy disease.
Motor nerve conduction study in ALS and ALS variants typically showed diffusely reduced CMAPs with no conduction blocks. Careful assessment of motor responses for demyelinating features (distal latencies >30% prolongation, conduction velocities <70% normal, and conduction blocks), which, if present, suggest an acquired demyelinating neuropathy such as multifocal motor neuropathy (MMN) and CIDP (10). Both MMN and CIDP may mimic ALS variants clinically. Needle EMG is the most important diagnostic study for ALS and ALS variants. It should demonstrate signs of active denervation with chronic denervation. Needle EMG is also crucial for delineating the pattern of myotomes involvement for regional ALS variants and excluding myopathy.
In addition to electrophysiologic study and neuroimaging, limited further testing may be required to exclude differential diagnoses in clinical scenarios of ALS or ALS variants. Transcranial magnetic stimulation (TMS) study may show prolonged central motor conduction times (CMCTs) despite the absence of pyramidal signs in ALS (11). To date, there is no data about TMS findings in regional ALS variants. Case 1 and case 2 in these case series had undergone TMS, and their CMCTs were normal.
Other differential diagnoses that can mimic regional ALS variants include Kennedy disease and monomelic amyotrophy (Hirayama disease). Male patients with a lower motor neuron syndrome with abnormal sensory responses on NCS should be tested for Kennedy disease with genetic testing for a CAG repeat expansion in the androgen receptor gene. Other diagnostic clues for Kennedy's disease include facial twitching, tremor, and features of androgen insensitivity (e.g., gynecomastia and testicular atrophy). Patients with monomelic amyotrophy usually presented with asymmetric hand and distal forearm weakness and atrophy with no sensory involvement, which may mimic BAD. The diagnostic clues to differentiate monomelic amyotrophy from BAD are younger age group and its self-limiting course.
BAD and LAD are unique regional variants of ALS with significantly better prognosis and survival than typical ALS. Understanding the striking phenotypic variation in these regional ALS variants may guide the prognosis of the disease. Recognizing these ALS variants is also important to exclude them in ALS clinical trials as it may obscure the effectiveness of therapy due to their slower progression.
CONCLUSION
BAD and LAD are unique regional variants of ALS with a significantly better prognosis than typical ALS. The phenotypic characteristics of regional ALS variants must be recognized when physicians are to tailor advice on disease progression, disease outcome, drug therapy, and end-of-life planning for patients with ALS or ALS variants.
REFERENCES
- Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG; Eurals Consortium 2009: Prognostic factors in ALS: A critical Amyotroph Lateral Scler 10:310-323.
- Ludolph A, Drory V, Hardiman O, et al. A revision of the El Escorial criteria-2015. Amyotroph Lateral Scler Frontotemporal Degener 2015; 16(5-6):291-292.
- Nodera H, Izumi Y, Kaji R. New diagnostic criteria of ALS (Awaji criteria). Brain Nerve 2007;59(10):1023- 1029.
- Chio A, Logroscino G, Traynor BG, et Global epidemiology of amyotrophic lateral sclerosis: a systemic review of the published literature. Neuroepidemiology 2013;41(2):118-130.
- Jawdat O, Satland JM, Barohn RJ, et al. Amyotrophic lateral sclerosis regional variants (brachial amyotrophic diplegia, leg amyotrophic diplegia, and isolated bulbar amyotrophic lateral sclerosis). Neurol Clin 2015;33(4):775-785.
- Ince PG, Evans J, Knoop M, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology 2003; 60(8): 1252-1258.
- Traxinger K, Kelly C, Johnson BA, et al. Prognosis and epidemiology of amyotrophic lateral sclerosis: analysis of a clinical population, 1997-2011. Neurol Clin Pract 2013;3(4): 313-320.
- Geevasinga N, Van den Bos M, Menon P, Vucic S. Utility of Transcranial Magnetic Simulation in Studying Upper Motor Neuron Dysfunction in Amyotrophic Lateral Sclerosis. Brain Sci. 2021 Jul 9;11(7):906. doi: 10.3390/brainsci11070906. PMID: 34356140; PMCID: PMC8304017.
- Collin Lauren E, et al. Amyotrophic lateral sclerosis and other motor neuron diseases. Continuum (Minneap Minn) 2020;26 (5, Peripheral nerve and motor neuron disorders): 1323-1347.
- Gwathmey K. Chronic inflammatory demyelinating polyradiculoneuropathy and its variants. Continuum (Minneap Minn) 2020; 26(5, Peripheral nerve and motor neuron disorders): 1204-1222.
- Kachi T, Sobue G, Yamada T, Tamura T, Ando Central motor conduction time in the pseudopolyneurotic form of amyotrophic lateral sclerosis. Rinsho Sjinkeigaku 1991;31: 1029-1031.